Ba’zan hayotda erkakshoda ayol va qizlar ham uchrab turadi. Hamma ularning qiz bola ekanligini biladi, lekin erkaksifat ko‘rinish va xususiyatlari sababli odamlar ulardan jinsiga nisbatan gumon qilib yurishadi. Ularning tashqi ko‘rinishi xuddi erkaklar kabi baquvvat muskullar va jismoniy holati bo‘yicha ham erkaklarga yaqin bo‘ladi. Bunday xususuyat ayollarda erkaklik garmon(testosteron)ining oshishi bilan tasniflanadi. Shuningdek, tibbiyotda ayollarda testosteron oshishidan tashqari ayollarni erkaksifat ko‘rinishiga sabab bo‘luvchi boshqa holatlar ham uchrab turadi. Xo‘sh, ayollarda bunaqa jins anomaliyasi holatlari qanday rivojlanadi? Buning asoslari nimada? Shu haqida ushbu maqolada batafsilroq to‘xtalib o‘tamiz.
Odamning jinsini belgilovchi genlar
Xromosomalar– hujayra yadrosining eng muhim tarkibiy qismidir. Xromosomalarda irsiy ma’lumotlarni saqlovchi va nasldan-naslga o‘tkazuvchi genlar joylashadi. Sochlar va ko‘zlarning rangidan tortib to odamning jinsini belgilovchi genlargacha bo‘lgan barcha genlar xromosomalarda joylashgan. Odamlarning erkak yoki ayol jinsga mansubligi ma’lum xromosomalarning mavjudligiga yoki yo‘qligiga bog‘liq. Odam hujayralarida 23 juft xromosoma mavjud bo‘lib, jami 22 juft autosomalar (jinsiy bo‘lmagan xromosomalar) va 1 juft jinsiy xromosomalar mavjud. Jinsiy xromosomalar har bir hujayradagi 23-juft xromosomalar hisoblanadi. Bular, X xromosoma va Y xromosomalardir. Har bir insonning har bir hujayrasi odatda bir juft jinsiy xromosomaga ega. Ayollarda odatda ikkita X xromosoma, erkaklarda esa bitta X va bitta Y xromosomalari mavjud. Bu juftlik jinsiy xromosomalar otaning urug‘ (sperma) hujayrasi va onaning tuxum hujayrasi qo‘shilishining o‘zidayoq odamning jinsini belgilaydi. Meyoz jarayonida erkaklardagi XY juft xromosomalari ajralib chiqadi va X yoki Y shaklida alohida gametalarga o‘tadi. Bu gametalarning shakllanishiga olib keladi, bunda hosil bo‘lgan gametalarning yarmi X xromosomasini o‘z ichiga oladi qolgan yarmi Y xromosomasini o‘z ichiga oladi. Spermatazoid X xromosomalik yoki Y xromosomalik bitta jinsiy xromosomani olib yuradi. Tuxum esa X xromosomasini olib yuradi. Shunga ko‘ra, agar tuxum X xromosomali spermatozoid bilan urug‘lantirilsa, embrionda ikkita X xromosoma (XX) bo‘ladi va agar tuxum Y xromosomali sperma bilan urug‘lantirilsa, embrion X va Y xromosomalarini o‘z ichiga oladi. Shuning uchun sperma va tuxumning birikmasi natijasida embrionning jinsi erkak (XY) yoki ayol (XX) sifatida aniqlanadi. XX xromosomalari bo‘lgan urg‘ochilar gomogametik, XY xromosomalari bo‘lgan erkaklar esa geterogametik jinslar deb nomlanadi.
Har doim ham XY erkak XX ayol jins bo‘lmaydi. Teskari holatlar ham bo‘lishi mumkin.
Y xromosomasida joylashgan SRY geni jinsni belgilovchi gen hisoblanib “Y proteini” deb ataladigan oqsilni sintez qilinishiga javob beradi. Bu oqsil erkaklarga xos jinsiy rivojlanishda ishtirok etadi. Bu protein homilada erkak jinsiy bezlar (moyaklar) rivojlanishiga olib keladigan va ayol jinsiy tuzilmalarining (bachadon va fallop naychalari) rivojlanishiga to‘sqinlik qiladigan jarayonlarni boshqaradi va bundan tashqari Y xromosomada immun tizimi ishlashini, erkak bo‘yini, ayrim tafakkur jarayonlarini boshqaruvchi genlar ham bor.
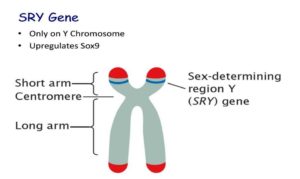
Jinsiy xromosomalar orasida genetik materialning anormal almashinuvi (translokatsiya) holatlari ham yuzaga kelishi kuzatiladi. Jinsiy xromosomalarda anormal almashinuv odamlarda sperma hujayralarining shakllanishi ya’ni spermatogenez paytida tasodifiy hodisa sifatida sodir bo‘ladi. SRY geni Y-xromosomaning uchida joylashgan bo‘lib, rekombinatsiya paytida SRY geni X xromosomasining bir qismiga ko‘chib o‘tib qolish holati (translokatsiya) sodir bo‘ladi. Agar homila o‘zida SRY genini saqlovchi X xromosomaga ega bo‘lgan sperma hujayrasidan homilador bo‘lsa, u Y xromosomaga ega bo‘lmasa ham, erkak sifatida rivojlanadi. XX erkak sindromi bo‘lgan shaxslarda erkak fenotipining rivojlanish darajasi, hatto SRY-musbat shaxslar orasida ham o‘zgaruvchan bo‘ladi. To‘liq erkak fenotipi odatda SRY geni ishtirokida rivojlanadi, lekin ba’zi hollarda SRY geni mavjudligi ichki yoki tashqi jinsiy a’zolarning noaniqliklariga olib kelishi mumkin. SRY geniga ega X xromosoma 90% hollarda faol bo‘lib, ko‘pincha SRY-musbat XX erkaklarda to‘liq erkak fenotipining rivojlantirishi kuzatiladi. SRY genisiz qolgan Y xromosoma saqlagan gameta X xromosoma bilan qo‘shilsa, XY ayol jins anomaliyasi(Svayer sindromi) rivojlanadi. SRY geni boshqa genlarning ekspressionini tartibga soluvchi transkripsiya omili bo‘lgan TDF oqsilini (moyakni belgilovchi omil) kodlaydi, o‘z navbatida embrional davrda erkak jinsiy tizimining rivojlanishini boshlaydigan transkripsiya omillarini kodlaydi. Meyoz paytida spermatogenez paytida Y xromosomasi ushbu genni X xromosomasiga o‘tkazish orqali SRY ni yo‘qotishi mumkin. Natijada, SRY genisiz bunday Y xromosomasining merosxo‘rligi Svayer sindromiga olib keladi
Svayer sindromi (to‘liq yoki sof gonadal disgenez- reproduktiv tizimning har qanday tug’ma rivojlanish buzilishi, embrionning rivojlanayotgan jinsiy bezlarida ibtidoiy jinsiy hujayralarning progressiv yo‘qolishi) qisqacha erkak genotipiga ega bo‘lgan ayol fenotipi sifatida tavsiflanishi mumkin. Kasallik ingliz endokrinologi Jerald Svayer sharafiga nomlangan bo‘lib, u 1955 yilda uni erkak psevdogermafroditizmi holati sifatida tasvirlagan. . Svayer sindromi bo‘lgan inson tanasida erkak tanasiga xos bo‘lgan xromosomalar to‘plami mavjud, ammo jinsiy bezlar (bir yoki ikkalasi) gonadaldir va gormonlar ishlab chiqarmaydi. Natijada, u ayol jinsiy a’zolariga, ayol jinsiy tizimiga ega va ayolga o‘xshaydi. Balog‘atga yetishish davrida ikkilamchi jinsiy xususiyatlarning rivojlanishi sodir bo‘lmaydi, psixologik rivojlanish ayol turiga qarab sodir bo‘ladi. Bunday bolalarda tashqi ko‘rinish va tashqi jinsiy a’zolar qizlarnikidek bo‘lgani sababli ular qizlar tarbiyasini olishadi. Qizlardek hayot kechirishadi, sevishadi va turmush qurishadi. Lekin turmush qurgach farzandli bo‘la olishmaydi, chunki ularda tuxum hujayralar va bachchadon rivojlanmagan bo‘ladi.
XX erkak va XY urg‘ochi jinsiy anomaliyalarning har biri 20 000 tug‘ilishdan taxminan 1 ta holatda uchraydi. 46 XX karyotipli fenotipik erkaklar odatda X xromosomasining qisqa qo‘liga ko‘chirilgan ma’lum Y xromosoma ketma-ketligiga ega. Xuddi shunday, 46 XY karyotipli ba’zi fenotipik ayollar jinsni aniqlash uchun mas’ul bo‘lgan Y xromosomasining hududini yo‘qotdilar. Ushbu shaxslarning 90 foizida sindrom Y-xromosomaning SRY geni tufayli yuzaga keladi. Ushbu sindrom turli aniqlash usullari orqali tashxis qilinadi va taxminan 1:20 000(20 mingta tug‘ilishdan 1 ta) yangi tug‘ilgan erkaklarda uchraydi, bu esa Klaynfelter sindromiga qaraganda kamroq tarqalgan.
Svayer sindromi kelib chiqish sabablari
Svayer sindromining etiologiyasi yaxshi o‘rganilmagan. Bugungi kunda ma’lumki, ko‘pincha patologiyaning paydo bo‘lishi Y xromosomasining qisqa qo‘lida joylashgan va asosan moyaklar shakllanishini nazorat qilish uchun javobgar bo‘lgan SRY genining yo‘qligi yoki mutatsiyasi bilan bog‘liq. Kasallikning oilaviy holatlarini kuzatishga asoslanib, hali noma’lum X-bog‘langan yoki autosomal genlar ishtirok etishi mumkin. Xavf omillari ham to‘liq aniqlanmagan. Umumiy mutagen ta’sirlardan (ionlashtiruvchi nurlanish va intoksikatsiya, virusli infektsiyalar, muvozanatsiz yoki kam ovqatlanish) va yuqorida aytib o‘tilgan irsiy yukdan tashqari, patologiya ehtimoli otaning yoshiga bevosita bog‘liq bo‘lishi mumkin deb taxmin qilinadi. Ko‘pincha homiladorlik paytida tanaga har qanday ta’sir va Svayer sindromining rivojlanishi o‘rtasidagi bog‘liqlikni aniqlashni hozirgi kunda imkoni mavjud emas.
Simptomlar
Ko‘pincha kasallik balog‘at yoshiga yetgandan so‘ng aniqlanadi. Balog‘at davrida Svayer sindromi balog‘atga yetish belgilarining yo‘qligi bilan tavsiflanadi. Qov va qo‘ltiq osti sohalarda faqat siyrak tuk o‘sishi kuzatilishi mumkin, lekin bu ko‘pincha o‘smaydi. Sut bezlari rivojlanmaydi yoki juda zaif rivojlangan bo‘ladi. Tuxumdon disgenezi bo‘lgan tana turi erkak kabi keng yelkali, katta ko‘krak qafasli va tor tos suyakli rivojlanadi. Svayer sindromi bo‘lgan ayollar ko‘pincha normal yoki o‘rtacha balandlikda, rivojlangan mushaklar va “og’ir” pastki jag‘ga ega. Ba’zida klitorisning yengil gipertrofiyasi paydo bo‘ladi, garchi odatda tashqi jinsiy a’zolar ayollarga xos rivojlangan. Bemorlar birlamchi bepushtlik, jinsiy aloqa yoki ginekologik tekshiruv vaqtida qinning etarli darajada rivojlanmaganligi sababli noqulaylik yoki og‘riq hissi haqida shikoyat qiladilar. Bu yilgi Parij olimpiyadasida katta muhokamalarga sabab bo‘lgan, ayollar boksi musobaqalarida genetik jihatdan erkak deb tamg‘a olgan ikki sportchilar: Jazoirlik Iymona Xalif va Tayvanlik Lin Yu Ting ko‘pchiligimizni yodimizda bo‘lsa kerak. Jamiyatda shunday e’tirozlarga sabab bo‘luvchi, XY ayol jins anomaliyasi har 20 mingta yangi tug‘ilgan chaqaloqlardan 1 tasida uchrar ekan.
Xulosa
Xulosa o‘rnida aytish mumkinki jinsni belgilovchi omilning bu faolligi tufayli jinsiy bezlarga aylanishi kerak bo‘lgan hujayralar rivojlanmaydi. Jinsiy hujayralarning rivojlanmaganligi natijasida jinsiy gormonlar chiqarilmaydi: na ayol, na erkak. Bu va boshqa gormonlar tanaga ta’sir qilmasa, homila ayol fenotipik rivojlanishni tanlaydi, tana ayol fenotipik turiga qarab rivojlanadi. Ayol fenotipining rivojlanishiga qaramay, tug‘ma gonadal disgenez (reproduktiv tizimning har qanday tug’ma rivojlanish buzilishi, embrionning rivojlanayotgan jinsiy bezlarida ibtidoiy jinsiy hujayralarning progressiv yo’qolishi) mavjud bo‘lib, u odatda kutilgan balog‘at davrida o‘zini namoyon qiladi. Bemorlarda fallop naychalari va jinsiy a’zolari bo‘lsa ham, ular hech qachon jinsiy bezlar moyak yoki tuxumdonga aylanmagan. Ushbu buzilishlar tufayli, o‘z navbatida, bachadon rivojlanishida nuqson bo‘ladi.
X.Sulaymonova nomidagi RESM Odam DNKsi sud-biologik ekspertizasi
laboratoriyasi eksperti S.H.Turdiboyev